
Drug approval in the healthcare markets EU vs. US
The healthcare sector contains within it, the essential pharmaceutical sub-sector that is tightly controlled by a set of processes and regulations, unique to each country. These regulations allow for the protection of each country’s citizens from harmful and unexpected consequences, potentially toxic drugs and damaging medical devices.
The industry has a responsibility to ensure the health and safety of the population by guaranteeing that pharmaceutical products are safe for public use and their efficacy outweighs any known or potential risks.
Product approval is exhaustive, lengthy, expensive and risky for corporates in order to provide the ‘guarantees’ alluded to above.
Globally, these guarantees fall primarily on the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) – the US and EU are the two most valuable pharmaceutical markets.
While the approval process is generally the same – research, design, clinical trials, regulatory approval, reporting and commercialisation – every country or region has its own regulatory authority. This means that even though a drug may be approved in the EU by the EMA and then authorised by the European Commission, it will not necessarily be approved in the US and vice versa.
In this blog we focus on the FDA and EMA given the US and EU are the largest pharmaceutical markets making up 49% and 23% drug manufacturing, respectively (IQVIA).
The US Food and Drug Administration (FDA)
The FDA was originally part of the Patent Office’s Agricultural Division commissioned in 1839. Its primary role was to collect statistics and report on agricultural developments.
After the US Department of Agriculture was founded in 1862, the division was transferred and became the Division of Chemistry in 1890 and consequently the Bureau of Chemistry in 1901.
In 1927 the Bureau’s name was changed to the US Food, Drug and Insecticide Administration and finally in 1930 its name was shortened to the US Food and Drug Administration.
Originally, the FDA was responsible for making sure that medications available to the public actually worked and that manufacturers did not embellish the benefits in their advertising.
Eventually, additional resources were injected into the agency to ensure that the drugs were not only efficient but also safe for the public use. Subsequently in 1976 the FDA was authorised to regulate medical devices.
FDA Approval Process
The FDA aims to approve the vast majority of drugs (over-the-counter and prescription – including biologics and generics) within six months of receipt of a drug approval application (exhibit 1). There is also a fast-track process – the FDA Accelerated Approval program for drugs required for life-threatening or unprecedented (e.g. Covid-19) conditions.
The FDA approval process is exceptionally rigorous – on average only five drugs in 5,000 that are part of pre-clinical trials are given the green light for human testing. Of those five generally only one is approved for sale.
The FDA approved 48 new drugs in 2019 vs. 59 in 2018.
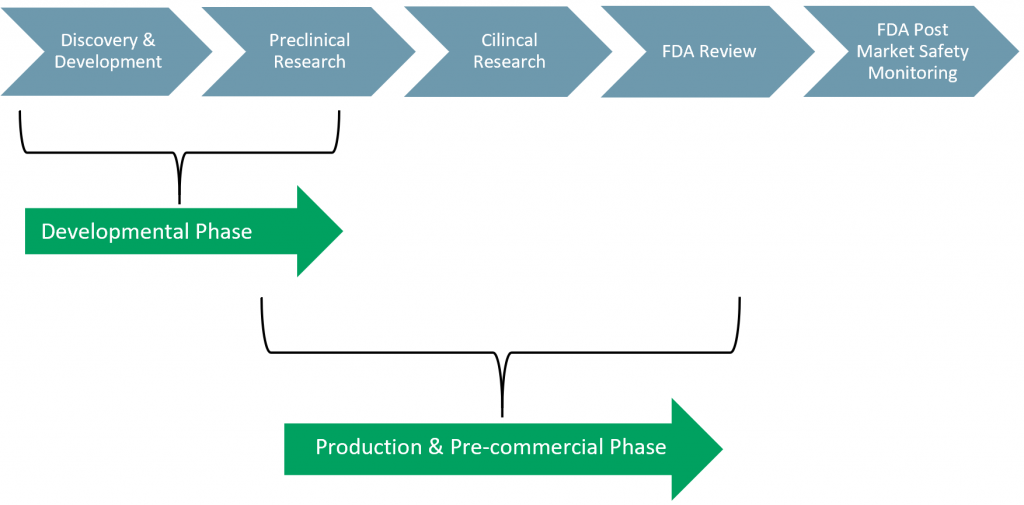
Exhibit 1 – FDA Approval Process
 Sources: ACF Equity Research; FDA
Sources: ACF Equity Research; FDA
The FDA approval process, for the sale of drugs, has five phases:
- Phase I: Discovery & Development
- The drug companies R&D process where the drug is developed in the lab.
- Phase II: Preclinical Research
- Laboratory and animal testing of the drug is done ‘In Vivo’ and ‘In Vitro’. Phase II generates preliminary statistics and test results to evaluate how humans might react to the drug.
- Phase III: Clinical Research
- Companies can begin the process for the Investigational New Drug (IND) application. The application must include stats of all lab and animal testing and an indication of how human testing will be carried out.
- Phase IV: FDA Review
- At this stage the FDA will begin to question the data and its interpretation – enter the Centre for Drug Evaluation and Research (CDER). The CDER does not only cover over-the-counter (OTC) drugs and prescriptions, but also everyday use toiletries, e.g. deodorant, anti-dandruff shampoo, sunscreen, and fluoride toothpaste.
- Once the IND is approved, companies can apply for a New Drug Approval (NDA) application.
- The NDA application can take several years and as indicated above only one in 1,000 drugs are approved at the pre-clinical trial stage. Once the NDA is approved, companies can begin their human clinical trials.
- Phase V: FDA Post-Market Safety Monitoring
- Following the approval process and successful completion of the applications, the FDA will continue to monitor said drug as there still could be issues once on the market. This allows the FDA to make any adjustments to dosage or usage.
FDA approval means that the “benefits of the product outweigh the known risks for intended use”.
The European Medicines Agency (EMA):
The EMA was created in 1995 replacing the Committee for Proprietary Medicinal Products and the Committee for Veterinary Medicinal Products. It is financed by the European Union, the pharmaceutical industry, and indirect subsidies from its member countries.
EMA is responsible for homogenising all the systems and processes throughout its member states regulatory organisations. It was set up to provide a more level and cost-effective playing field for pharmaceutical companies that previously were required to gain a different approval from each EU country. The agency was also able to restrict regulations that were fostering competition amongst the member states.
EMA has two broad approaches to the approval processes – centralised and decentralised
EMA’s centralised approval system – provides a centralised approval process (submission of a single application) where approved drugs are then authorised by the European Commission for marketing in all EU and non-EU EEA countries. The EMA also provides a centralised monitoring system for drug safety, and product information is made available in all EU languages.
It is compulsory for the following categories of drugs to go through the EMA centralised approval process – biotech processed drugs, HIV/AIDS and other auto-immune and immune dysfunctions, cancer, diabetes, neurodegenerative diseases, viral diseases, gene-therapy, somatic cell-therapy, tissue-engineered, orphan status, and veterinary medication for growth or yield enhancement (e.g. antimicrobial agents).
EMA’s decentralised system – allows for drugs to be approved in more than one EU member state simultaneously – so multiple approvals but not every state at once. The additional advantage is that it can be used for drugs that do not need to be authorised under the centralised system (i.e. EMA).
In our view, the National Procedure (NP) and Mutual Recognition procedures (MRP) essentially fall under a decentralised system approach.
The NP approach is best used when the goal is to gain approval in a single EU member state as a stepping stone, but not that each EU member state has its own national procedure.
The MRP approach allows an NP authorization to be recognised in other EU member states and is a stepping stone approach to potentially gain authorisation in more or all other EU member states.
EMA Approval Process
The EMA drug approval process is less rigorous than that of the FDA and on average takes about two months (Exhibit 2). In 2019, the EMA approved 66 new drugs of which 30 had new active substances previously never authorised vs. 84 new drug approvals in 2018.
A European pharmaceutical company can seek approval through five different routes, which does not limit it to the EMA. The manufacturer’s choice of route is determined by several factors, e.g. the type of drug, application preference and markets at which the drug is targeted.
The routes are:
- EIND = Emergency Investigational New Drug;
- EMA = European Medicines Agency;
- EU = European Union;
- FDA = Food and Drug Administration;
- IND = Investigational New Drug.
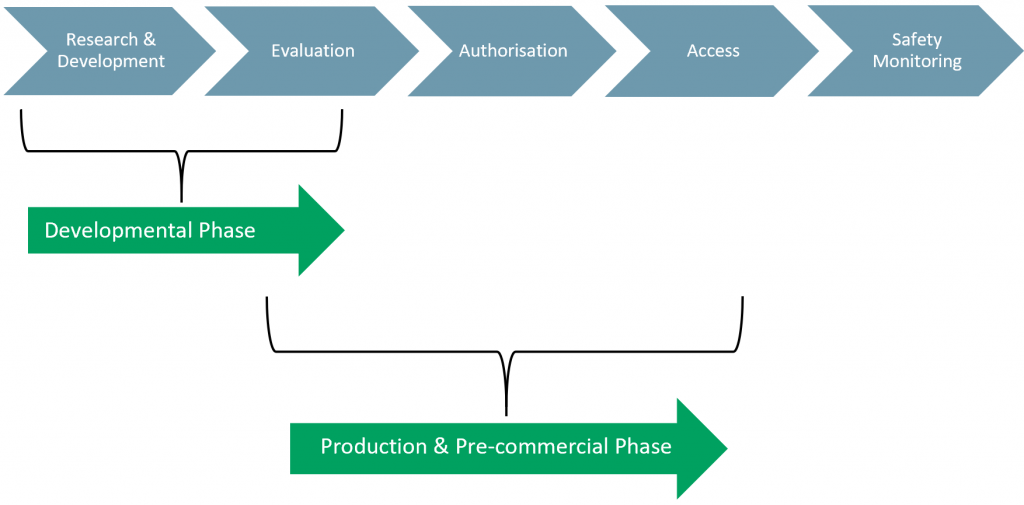
Exhibit 2 – EMA Approval Process
 Sources: ACF Equity Research; FDA
Sources: ACF Equity Research; FDA
Like the FDA, the EMA approval process also has five phases:
- Phase I: Research & Development
- The drug companies R&D process where the drug is developed in the lab.
- Phase II: Evaluation
- Companies supply data and request authorisation. EMA begins its assessment and prepares questions and may consult experts during its investigation.
- Phase III: Authorisation
- The EC takes the final decision on whether or not the drug or device can be marketed in the EU.
- Phase IV: Access
- National and regulatory authorities decide on pricing and reimbursement.
- Phase V: Safety Monitoring
- Once a drug is authorised, EMA and national authorities continue to monitor its safety.
Any drug or “any substance or combination of substances which may be administered to human beings or animals with a view to making a medical diagnosis or to restoring, correcting, or modifying physiological functions in human beings or in animals” is eligible for EMA approval/authorisation.
As the EMA does not consider it essential to demonstrate the efficacy of a drug, a lot more applications are generally successful during the early stages of the process compared with the US process run by the FDA.
The FDA vs. EMA
The main difference between the two agencies is the approval timeline. The FDA’s process is, overall, notably slower and so more expensive, than the EU’s EMA process. However, the FDA’s Phase IV Review is shorter than EMA’s equivalent Phase II Evaluation.
This difference in time spent on an ‘in-depth’ analysis and review of a new drug is the topic of many conversations around the integrity of the agency’s processes. However, voting for accuracy of evaluation/review vs. speed of approval is not so black and white. As we have learned from the Covid-19 pandemic, speed of approval literally is a matter of life and death.
Another difference is attributed to the clarity and transparency of the processes. The lack of transparency of clinical trial data is a cause of concern for both agencies as they struggle to produce consistent reviews and statistical analysis of drugs.
While both the FDA and EMA approval processes are efficient from a safety perspective, it is often difficult to assess which one is better from a commercial perspective. The drug approval process for the US and EU would be better if it were homogenized.
In our view, homogenisation of the drug approval process across the US and EU markets would reduce the cost to bringing drugs and devices to market and reduce the risks faced by investors. The reduced cost and risk of drug approval would lower the internal rate of return (IRR) for a new drug.
A lower IRR for drug development would lead to more drugs coming to market. More drugs coming to market, ceteris paribus, would increase patient well-being and competition. Greater competition increases the overall value of a market and reduces the cost of drugs and devices to the ultimate payees (insurers and national healthcare systems).












{kind=link}